Personalized Vaccines Coming Soon?
Hana Dobrovolny’s research could lead to antiviral drugs.
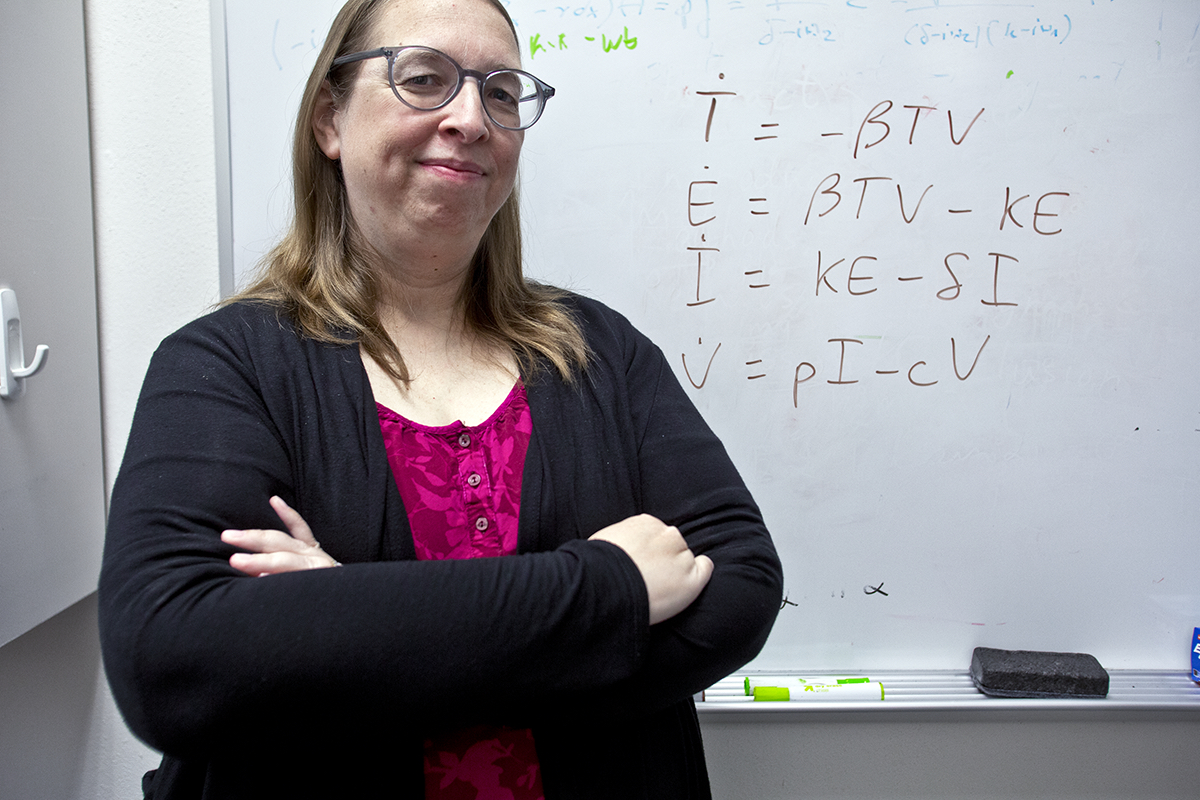
Photo by Mark Graham
Personalized Vaccines Coming Soon?
Hana Dobrovolny’s research could lead to antiviral drugs.
In 1918, tens of millions of people worldwide died in one of the largest modern pandemics. The culprit was a variant of the H1N1 influenza virus, a genetic relative of many of the 21st century’s flu strains. Scientists in the early 20th century didn’t fully understand viruses, let alone how they mutate. Those small genetic changes are, in part, why people need a vaccination every year and why the flu develops resistance to antiviral drugs.
Leading the battle against flu are public health authorities who rely on computational experts like Hana Dobrovolny, associate professor of physics and astronomy. She determines how influenza moves and mutates through the body so researchers can develop antiviral drugs.
“The flu is the fastest-mutating virus,” Dobrovolny said. “Within the first 24 hours, you have had every single amino acid” mutated. After two or three days, the virus will have completely mutated twice, and as Dobrovolny said, “This is why drug resistance is easy.”

A laptop screen displays visualizations of the results of recent studies in influenza drug resistance conducted by Hana Dobrovolny, associate professor of physics and astronomy. Photo by Mark Graham
Only two drugs currently target influenza: neuraminidase inhibitors such as Tamiflu and adamantanes such as Flumadine, which are no longer recommended in the U.S. Most strains of the flu are resistant to all adamantanes — the first of these was developed in 1966, and the most recent adamantane was developed in 1994. Complete resistance was observed in the 2008-2009 flu season. Newer drugs are variants of neuraminidase inhibitors.
The two types of flu-fighting drugs function at different points in the virus life cycle. Think of a virus as having docking proteins and undocking proteins. The cycle begins with the incoming docking protein, hemagglutinin, attaching to a receptor on the surface of the cell. Once the virus is docked, it enters the cell and delivers its genetic material. That genetic material is transcribed within the cell’s nucleus, becoming instructions for building a copy of the virus that infected the cell. Finally, the neuraminidase protein undocks the newly formed virus, releasing it to find a new cell to infect.
Antiviral drugs latch onto the docking proteins, in the case of adamantanes, or the undocking proteins, in the case of neuraminidase inhibitors. When mutations occur in either of these proteins, the antiviral drug no longer works.
Adamantanes work at the beginning of the virus life cycle to stop replication. Neuraminidase inhibitors work at the end of the life cycle, stopping viral release. However, Dobrovolny said, “Both antiviral drugs need to be taken within the first 48 hours of infection, but most people do not realize they have the flu until after that.”
Computer models of viral activity within and between cells allow researchers to predict how infected cells are likely to respond to an untested drug. The data from these models helps researchers know which drugs to pursue in clinical settings.
“Models can give an upper limit and lower limit of a drug dosage that can then be explored clinically,” Dobrovolny said. “The models are not good enough to make predictions for a single person. But they set boundaries.”
A computer model can run through hundreds of calculations involving drug combinations, dosages and courses of treatment in minutes. Those models also help scientists see when drug resistance occurs and identify possible ways to overcome it.
Dobrovolny’s research uses agent-based models to look at the way flu moves around the body. These models, unlike typical computational models, do not start with an inference from the data. Instead, agent-based models apply rules to a system and see what happens. If results match known data, researchers can use the same rules and mechanism to look at unknown situations, such as a new drug or a new environment.
“The flu is the fastest-mutating virus. … This is why drug resistance is easy.”
Hana Dobrovolny
Dylan Barth ’18 worked on these rule-based mathematical models in Dobrovolny’s lab. “Our agent-based model of influenza was used to investigate the cell-to-cell infection dynamics of the influenza virus and also to model superinfection: a model of infection where two pathogens may infect the same cells or hosts,” he said.
The goal was to observe how the flu virus travels between cells and how the virus infects cells that are far away from the first infected cell, he said. “The interplay of these infection strategies comprises the larger story of intracellular infection.”
Baylor Fain, a graduate student in physics who took over the agent-based model project, said that after graphing initial results, the researchers found the rate at which the virus moved throughout a group of cells particularly interesting.
As it turns out, the way the flu moves from one cell to another depends on whether it interacts on the periphery of the cell through the surface proteins or goes directly from cell to cell through a nanopore tunnel. This affects how quickly the cell neighborhood becomes completely infected.
In 2008, researchers at the National Institute of Allergy and Infectious Diseases analyzed lung samples and autopsies from the 1918 flu pandemic and found evidence of a respiratory co-infection. This bacterial infection is like many co-infections today. In the 21st century, bacterial and viral infections are treated with different types of drugs, which means treatment might need to be patient-specific.
Dobrovolny said individualized treatment is the next step in computational studies of influenza. “The ultimate goal of all modelers is personalized models with all the details — the immune response, how the drug resistance will arise — that is specific for each person.”
COVID-19 Modeling
Long before the coronavirus outbreak, Hana Dobrovolny’s graduate student Lubna Pinky ’16 MS (PhD ’18) had developed a model to predict the molecular process of viral co-infection. Using available coronavirus data, the duo ran the model to show what might happen if the novel pathogen infected someone also nursing a more traditional virus. “It turns out that coronavirus is pretty weak,” Dobrovolny said, noting that flu and a common cold would suppress coronavirus. The bad news: If someone gets COVID-19 and then the flu, the model shows the latter would rage before the former, Dobrovolny said, and “hospitals could expect a double peak.” The model shows potential good news for people who fall prey to a more common virus: “If you get flu first, there’s almost no chance you can get a co-infection.”
— Caroline Collier
Your comments are welcome
Comments
Related reading:
Mapping the Coronavirus
Emma Hodcroft says warmer weather may slow the virus, but that doesn’t mean it’s contained.
Research + Discovery
Floyd Wormley Jr. Leads Research and Graduate Studies
The associate provost, dean and professor works to perfect the teacher-scholar model and help faculty learn to market their research.
Research + Discovery
Learning to Avoid Risky Behavior
Probationers use an app developed at the Institute of Behavioral Research to model better decision-making.